BLD Insights
Application of Bicyclic Pyrrolidine in Drug Development
18 January 2022
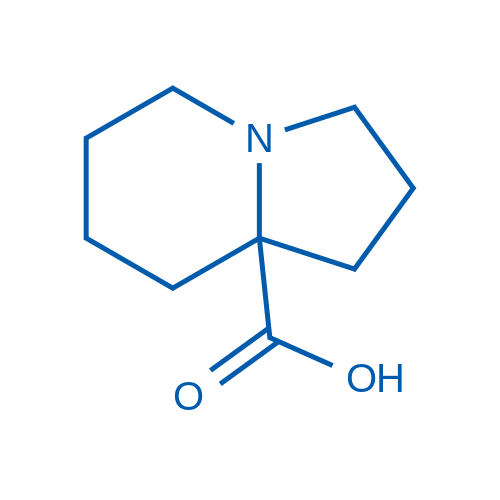
Octahydroindolizine-8a-carboxylic acid
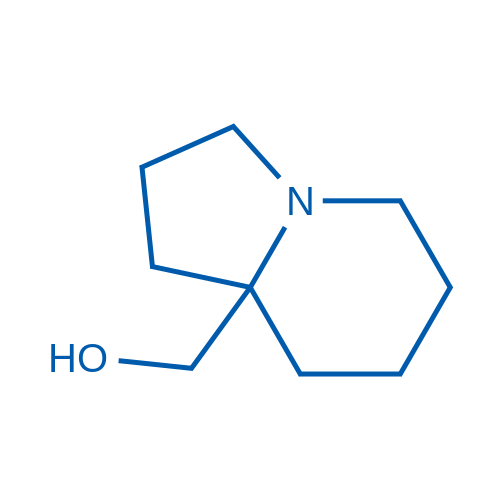
(Hexahydroindolizin-8a(1H)-yl)methanol
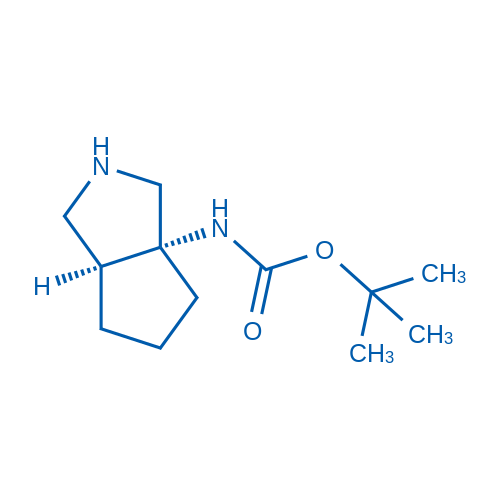
rel-tert-Butyl ((3aR,6aS)-octahydrocyclopenta[c]pyrrol-3a-yl)carbamate
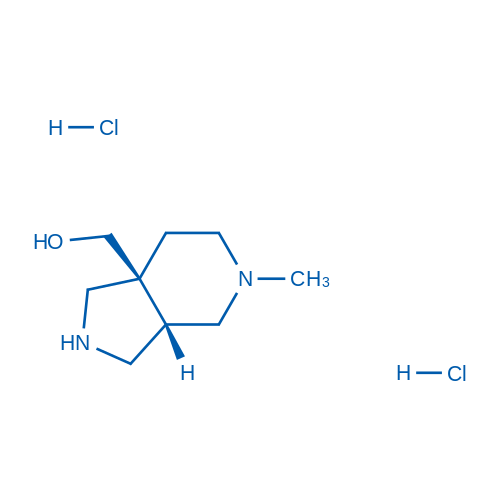
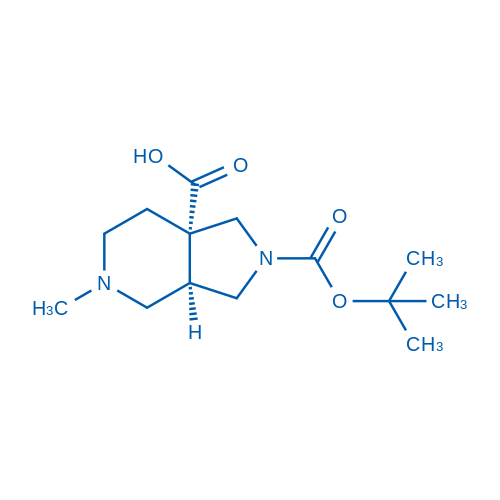
rel-(3aS,7aS)-(5-Methyloctahydro-7aH-pyrrolo[3,4-c]pyridin-7a-yl)methanol dihydrochloride
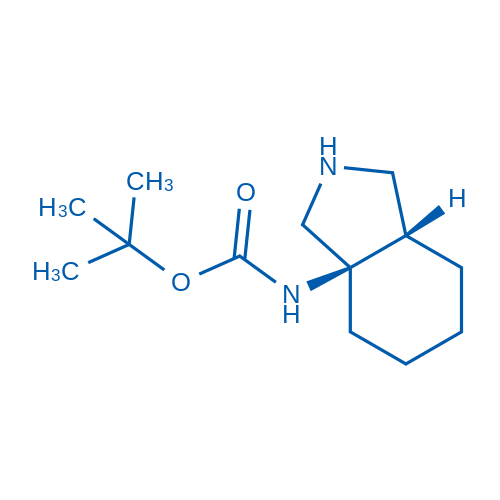
rel-tert-Butyl ((3aR,7aS)-octahydro-1H-isoindol-3a-yl)carbamate
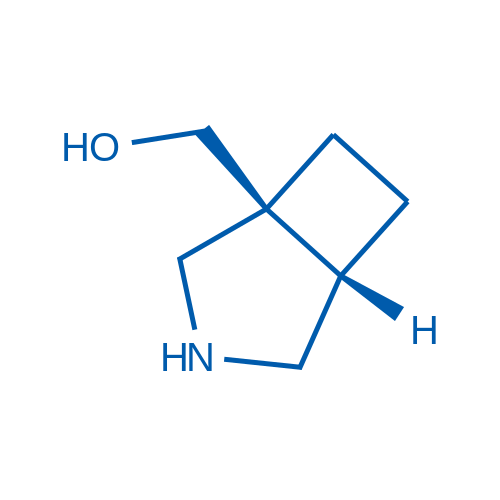
rel-((1S,5S)-3-Azabicyclo[3.2.0]heptan-1-yl)methanol
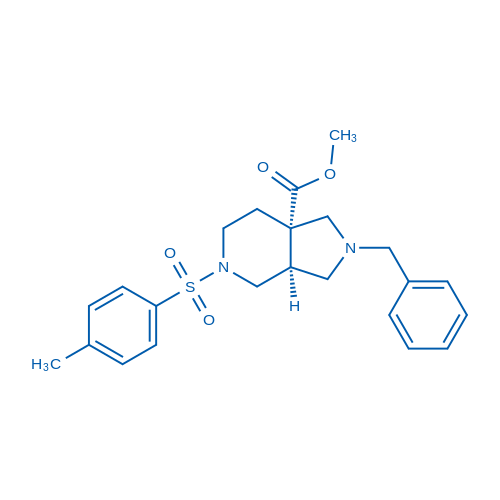
rel-Methyl (3aS,7aS)-2-benzyl-5-tosyloctahydro-7aH-pyrrolo[3,4-c]pyridine-7a-carboxylate
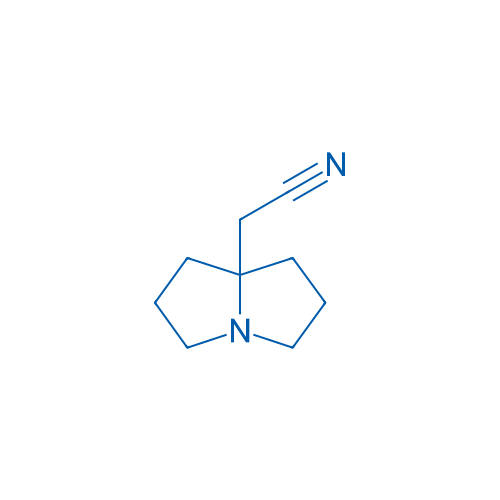
2-(Hexahydro-1H-pyrrolizin-7a-yl)acetonitrile
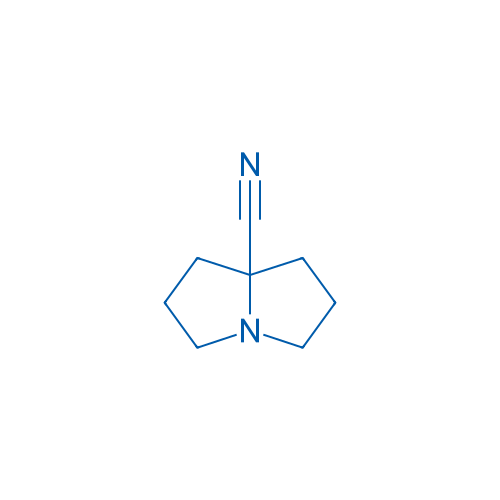
Hexahydro-1H-pyrrolizine-7a-carbonitrile
Octahydroindolizine-8a-carboxylic acid
(Hexahydroindolizin-8a(1H)-yl)methanol
rel-tert-Butyl ((3aR,6aS)-octahydrocyclopenta[c]pyrrol-3a-yl)carbamate
rel-(3aS,7aS)-(5-Methyloctahydro-7aH-pyrrolo[3,4-c]pyridin-7a-yl)methanol dihydrochloride
Nitrogen heterocycles are one of the most significant structural components of pharmaceuticals. 59% of unique small-molecule drugs contain a nitrogen heterocycle, among FDA-approved drugs. In the category of drugs containing five-membered nonaromatic nitrogen heterocycles, pyrrolidine is the most frequently used core (Fig. 1), which has become one of the most commonly used secondary amines in organic synthesis and medicinal chemistry [1]. Among them, bicyclic pyrrolidines play a unique role in numerous bioactive naturally occurring compounds and pharmaceutical ingredients (Fig. 2) [2].
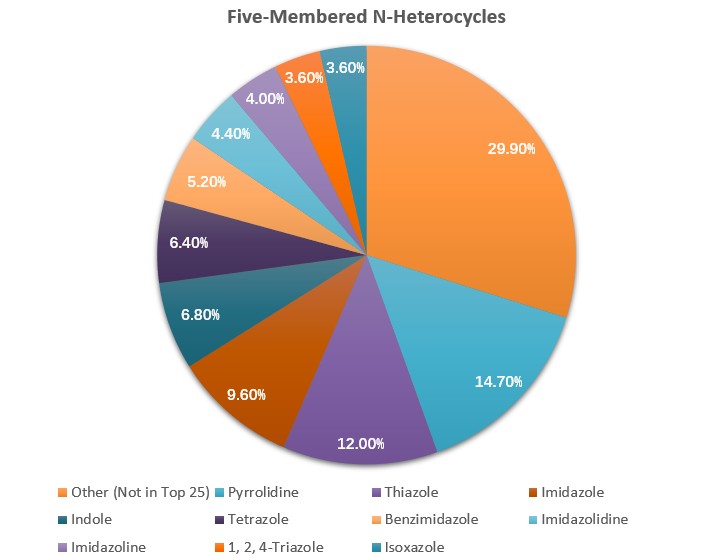
Figure 1. Distribution of five-membered nitrogen heterocycles.
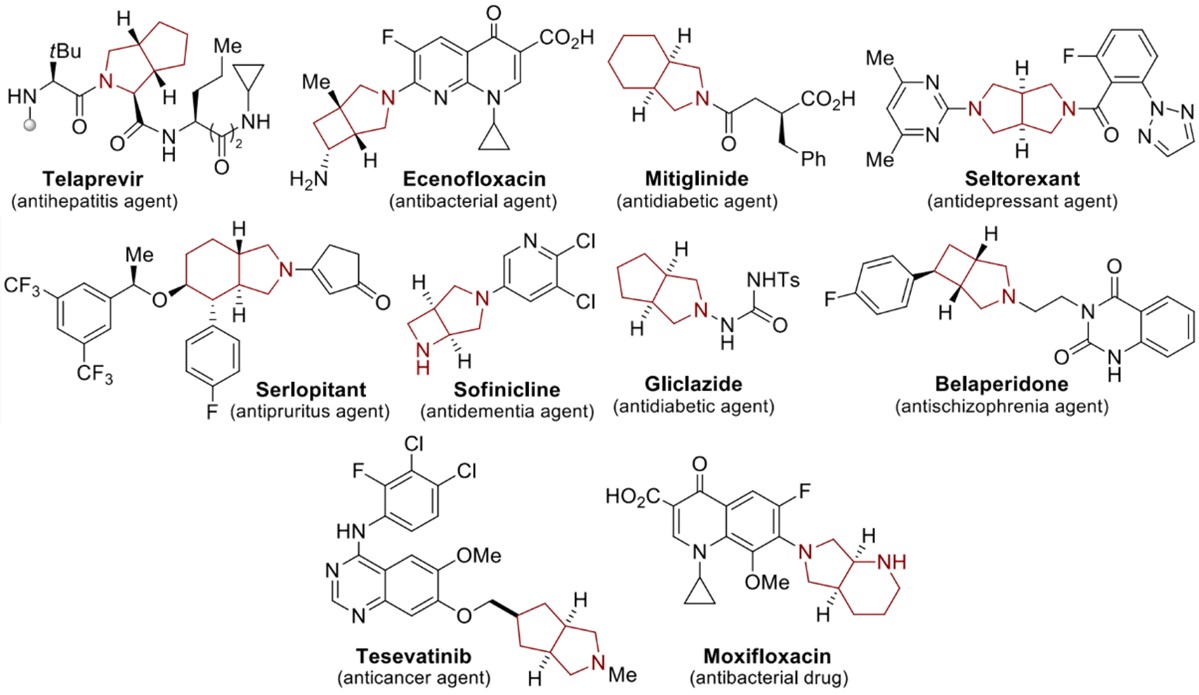
Figure 2. Bicyclic pyrrolidines in drug discovery.
Advantages of bicyclic pyrrolidine in drug design
Modern drug evolves rapidly, and researchers have increasing interests in scaffold hopping, escape flatland, and conformational restriction to expand the structure and improve the potency. Scaffold hopping aims to discover structurally novel compounds starting from known active compounds by modifying the central core structure of the molecule, which are built up through different synthetic routes and have new pharmacological properties [3]. Escape flatland is a way to increase the molecules' saturation to improve the probability of success in compound translation to the clinic. Fsp3 (number of sp3 hybridized carbons/total carbon count) is used to measure the complexity of the compound. A higher fraction of sp3 has an increased probability of success in compound translation to the clinic, and it may be mainly due to out-of-plane substituents, which is conducive to adjusting the molecular shape and increasing receptor/ ligand complementarity [4]. For another, receptor-ligand binding leads to entropy reduction. To overcome such entropy penalties and improve the potency of a ligand, a suitable strategy is to restrict its conformational flexibility. Therefore, the conformational restriction is of high interest to medicinal chemists [5]. The introduction of bicyclic pyrrolidine is also in line with these concerns, which combines alkane and pyrrole, is a 3D-shaped Fsp3-rich building block, and is intrinsically conformationally restricted. For example, substituted 3-azabicyclo [3.2.0]-heptanes are introduced as conformationally restricted surrogates for common piperidine motifs to provide 3D characteristics and increase conformational restrictions (Fig. 3). Compared with piperidine, 3-Azabicyclo [3.2.0]-heptane has nearly identical lipophilicity (logD), water solubility (Sol.), and metabolic stability (CLint) (Fig. 4), which is good a conformationally restricted surrogate for piperidine needed in modern medicinal chemistry projects [6].
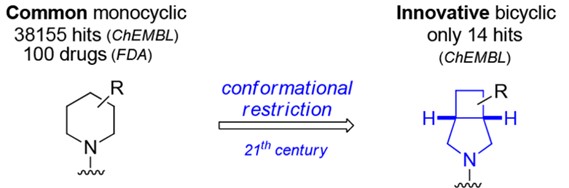
Figure 3. Piperidine and 3-azabicyclo [3.2.0] heptane.
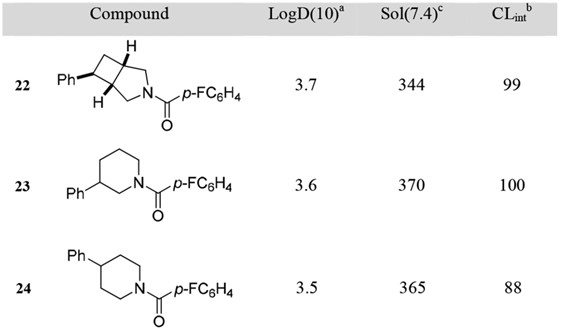
Figure 4. Measured Physicochemical Properties of piperidine and 3-azabicyclo [3.2.0] heptane.
At present, many drugs contain bicyclic pyrrolidine, here is a brief introduction with the development of Telaprevir as an example. Telaprevir, HCV NS3-4A protease inhibitor, has been approved for the treatment of genotype 1 chronic HCV in the United States, Canada, European Union, and Japan [7]. NS3-4A protease is a serine protease with a chymotrypsin-like fold, a perspective target for drug development. But, the protease active site is extremely hydrophobic, flat, and exposed to solvent, with few pockets to increase the affinity for an inhibitor to bind. The initial decamer peptide inhibitor was derived from the natural NS5A-5B substrate (Fig. 5(1)) [7]. To limit the molecular weight, the decamer inhibitor was truncated to subsites P4 to P1ʹ, despite the loss in potency. [8]. To compensate and increase affinity, Perni et al. developed a reversible covalent inhibitor, (Figure. 5(2)) [9]. Replacement of the aldehyde warhead with an α-ketoamide, increased ionic interactions, lengthened half-life, and improved binding affinity (Figure. 5(3)) [10]. Replacement of α-ketoamide with bicyclic ketone further improved the binding affinity of the substrate and the protein (Figure. 5(4)) [11]. Comprehensive optimization at the P1ʹ, P1, P3, and P4 binding subsites to reduce the ketone led to the synthesis of telaprevir (Fig. 6) [12]. In this case, the introduction of the bicyclic pyrrolidine greatly improved the binding affinity and facilitated the success of drug development.
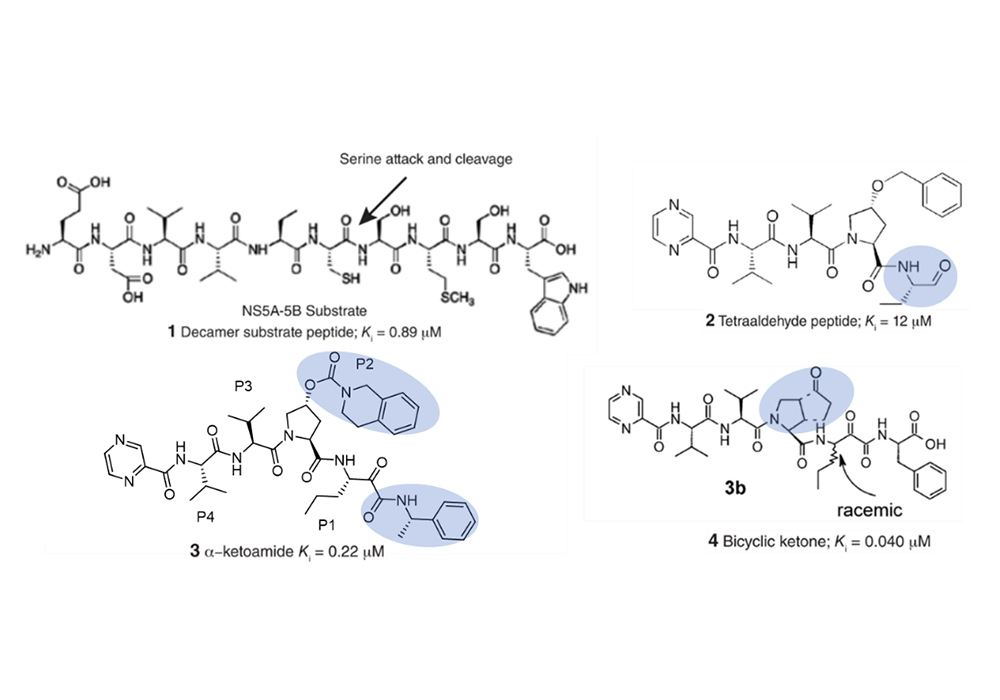
Figure 5. Evolution of NS3-4A inhibitor (1)
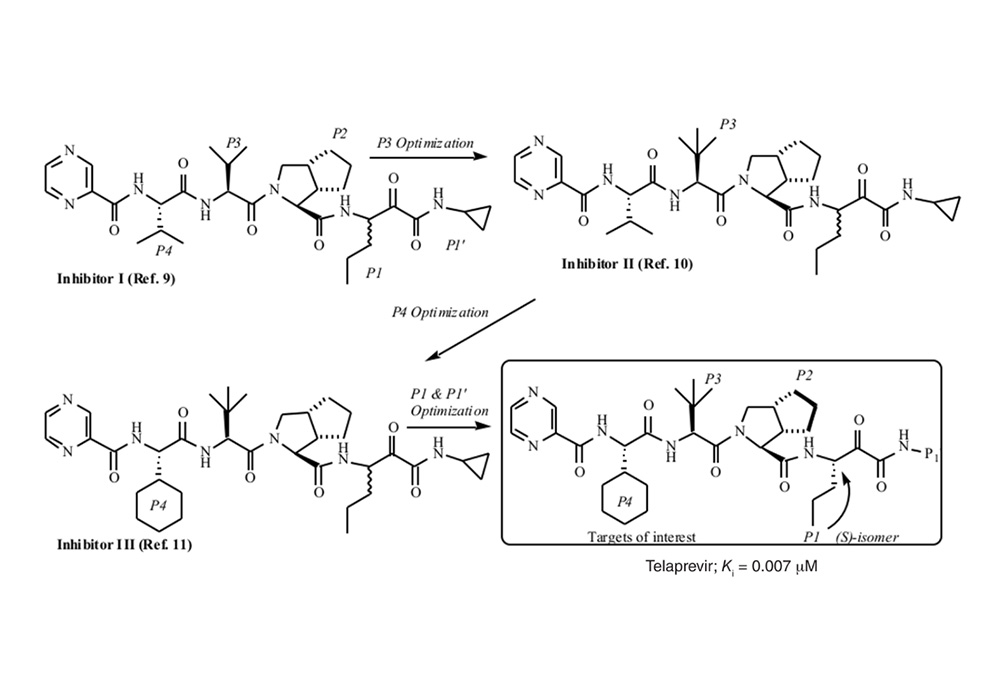
Figure 6. Evolution of NS3-4A inhibitor (2).
References
[1]Vitaku, E., Smith, D.T., and Njardarson, J.T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA-approved pharmaceuticals. J Med Chem 57, 10257-10274.
[2]Savych, V.I., Mykhalchuk, V.L., Melnychuk, P.V., Isakov, A.O., Savchuk, T., Timoshenko, V.M., Siry, S.A., Pavlenko, S.O., Kovalenko, D.V., Hryshchuk, O.V., et al. (2021). Bicyclic Pyrrolidines for Medicinal Chemistry via [3 + 2]-Cycloaddition. J Org Chem 86, 13289-13309.
[3]Bohm, H.J., Flohr, A., and Stahl, M. (2004). Scaffold hopping. Drug Discov Today Technol 1, 217-224.
[4]Lovering, F., Bikker, J., and Humblet, C. (2009). Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem 52, 6752-6756.
[5]Mann, A. Conformational restriction and/or steric hindrance in medicinal chemistry. In The Practice of Medicinal Chemistry, 3rd ed.
[6]Denisenko, A.V., Druzhenko, T., Skalenko, Y., Samoilenko, M., Grygorenko, O.O., Zozulya, S., and Mykhailiuk, P.K. (2017). Photochemical Synthesis of 3-Azabicyclo[3.2.0]heptanes: Advanced Building Blocks for Drug Discovery. J Org Chem 82, 9627-9636.
[7]Kwong, A.D., Kauffman, R.S., Hurter, P., and Mueller, P. (2011). Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat Biotechnol 29, 993-1003.
[8]Attwood MR, B.J., Campbell AD, Canning GG, Carr MG, Conway E, Dunsdon RM, Greening JR, Jones PS, Kay PB, Handa BK, Hurst DN, Jennings NS, Jordan S, Keech E, O'Brien MA, Overton HA, King-Underwood J, Raynham TM, Stenson KP, Wilkinson CS, Wilkinson TC, Wilson FX. (1999). The design and synthesis of potent inhibitors of hepatitis C virus NS3-4A proteinase. Antiviral Chemistry & Chemotherapy 10, 259-273.
[9]Perni, R.B., Britt, S.D., Court, J.C., Courtney, L.F., Deininger, D.D., Farmer, L.J., Gates, C.A., Harbeson, S.L., Kim, J.L., Landro, J.A., et al. (2003). Inhibitors of hepatitis C virus NS3.4A protease 1. Non-charged tetrapeptide variants. Bioorg Med Chem Lett 13, 4059-4063.
[10]Perni, R.B., Pitlik, J., Britt, S.D., Court, J.J., Courtney, L.F., Deininger, D.D., Farmer, L.J., Gates, C.A., Harbeson, S.L., Levin, R.B., et al. (2004). Inhibitors of hepatitis C virus NS3.4A protease 2. Warhead SAR and optimization. Bioorg Med Chem Lett 14, 1441-1446.
[11]Yip, Y., Victor, F., Lamar, J., Johnson, R., Wang, Q.M., Barket, D., Glass, J., Jin, L., Liu, L., Venable, D., et al. (2004). Discovery of a novel bicycloproline P2 bearing peptidyl alpha-ketoamide LY514962 as HCV protease inhibitor. Bioorg Med Chem Lett 14, 251-256.
[12]Lampa A, E.A., Vema A, Åkerblom E, Lindeberg G, Danielson UH, Karlén A, Sandström A. (2011). P2-P1' macrocyclization of P2 phenylglycine based HCV NS3 protease inhibitors using ring-closing metathesis. Bioorg Med Chem Lett 19, 4917-4927.