BLD Insights
A General ADC Linker - vcMMAE
29 April 2022
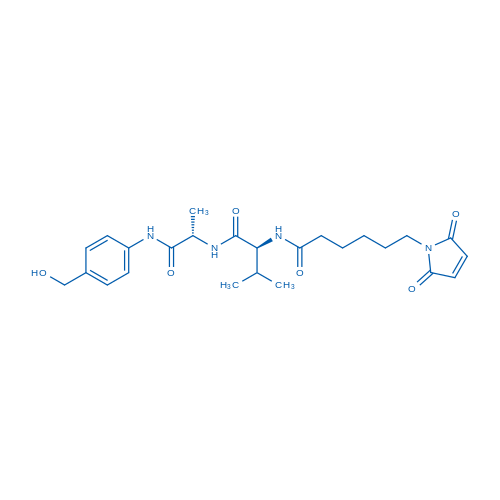
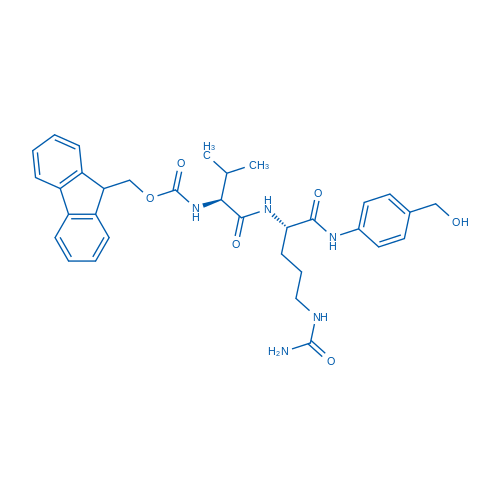
Val-Cit-PAB-OH
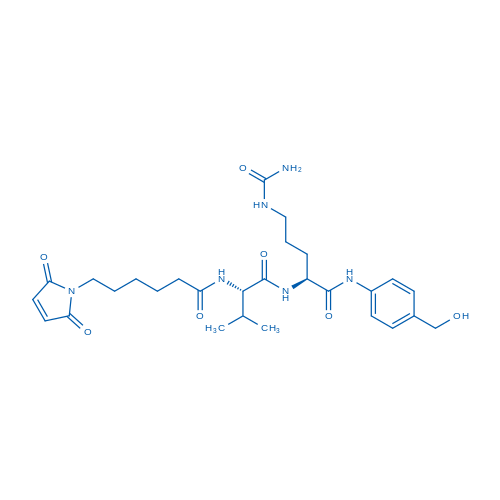
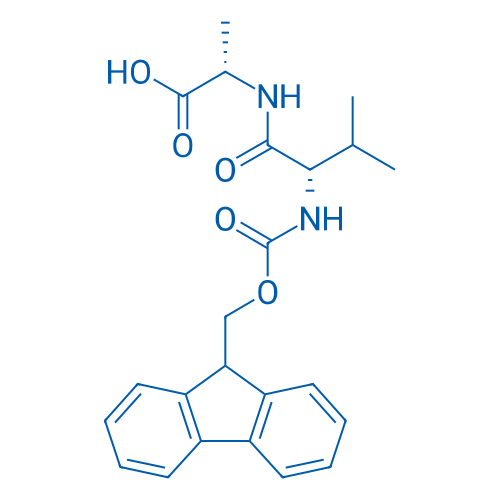
Fmoc-Val-Cit-PAB-OH
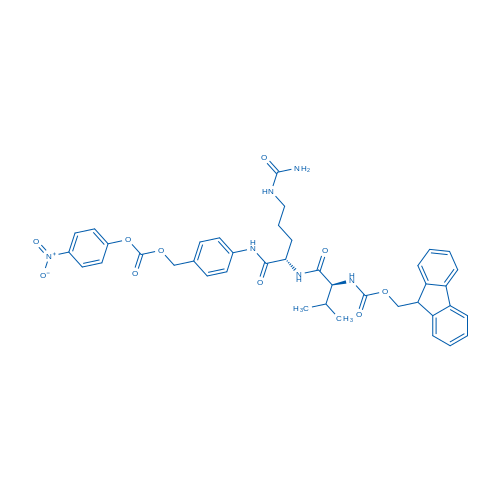
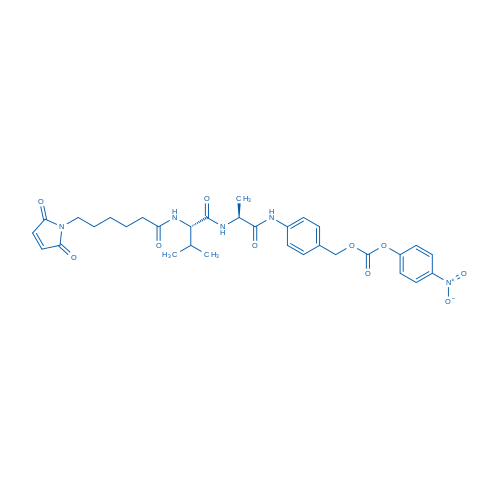
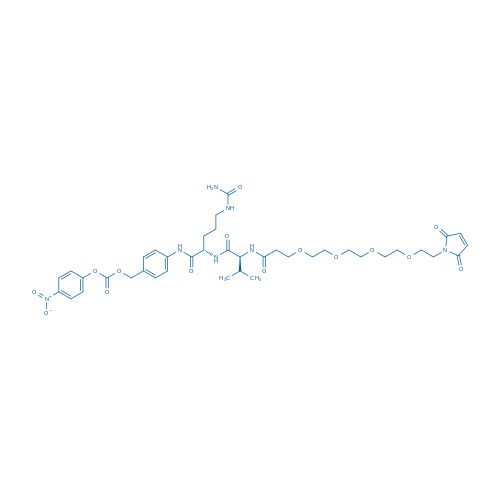
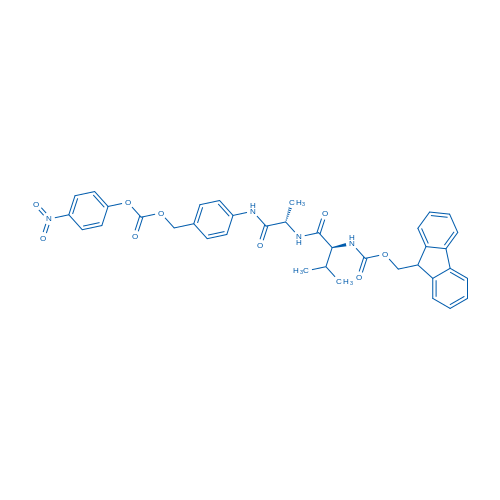
Fmoc-Val-Cit-PAB-PNP
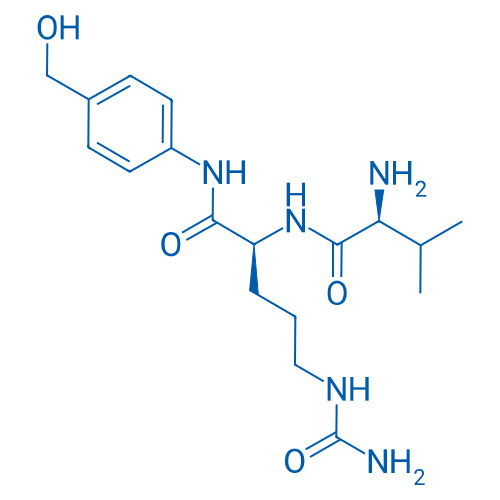
Val-Cit-PAB-OH
Introduction of Antibody-Drug Conjugate(ADC)
In recent years, monoclonal antibody(mAb) based reagents have shown considerable effectiveness in the clinical treatment of cancer. One of them, antibody-drug conjugates(ADC), can enhance drug efficacy through targeted delivery while sparing nontarget tissues from chemotherapeutic damage, thus becoming a popular therapeutic approach.
Most of ADCs share a similar mechanism of action. First, they will bind to specific antigens via their antibody, which leads to endocytosis of the cancer cell. Then, ADC molecules are transported from early endosomes to lysosomes for degradation of the foreign matter[1]. During this process, common ADC molecules are capable of cleavage between the antibody and the payload(a kind of cytotoxic small molecule), which is achieved by a specially designed cleavable linker. Finally, the payload exerts cytotoxic function after diffusing out of the endosome(Figure 1). In the early years, cleavable linkers adopted a hydrazone link for selective hydrolysis inside tumor cell lysosomes[2]. Besides its limited hydrolytic stability, the hydrazone could be used to carry only ketone-containing drugs such as DOX or its close analogs[3]. Therefore, scientists sought to replace the hydrazone with cathepsin B-sensitive peptides as they can be used to deliver a greater variety of drugs due to cathepsin B being never found extracellularly, except in pathological conditions such as metastatic tumors or areas of tissue destruction. As a result, conjugates produced with cathepsin B-cleavable linkers are likely to be stable in circulation.
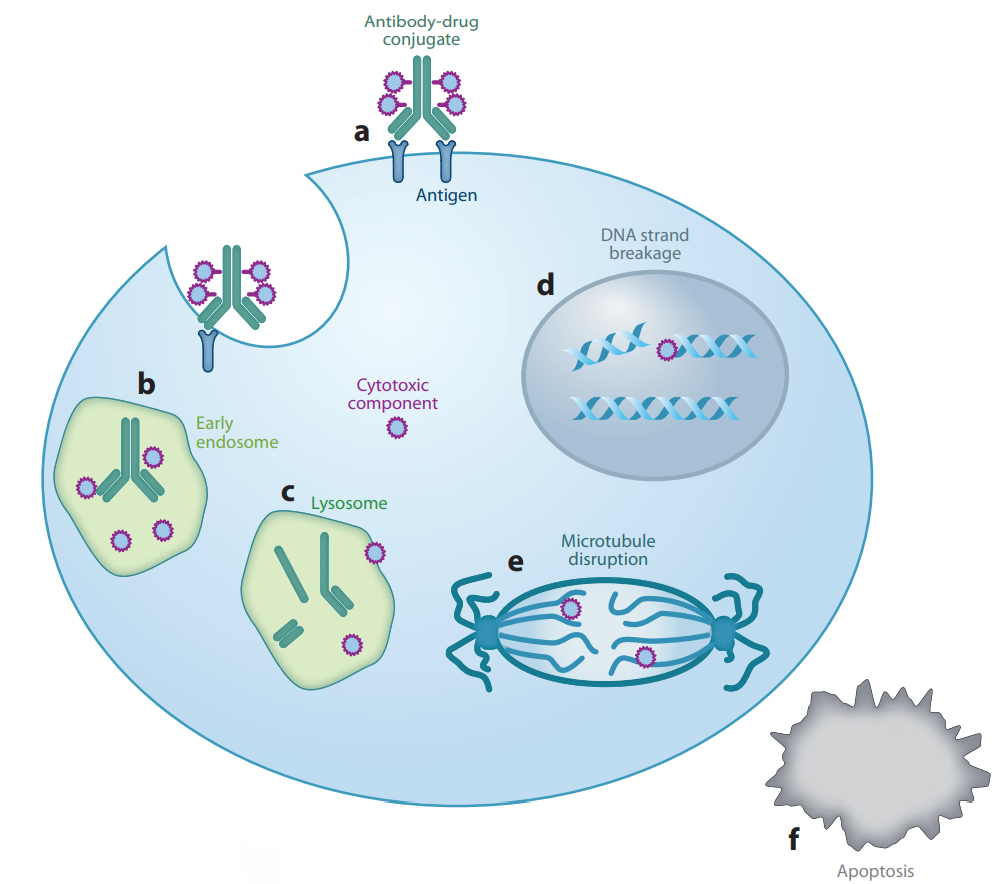
Figure 1: Generalized mechanism of action of ADC. a. antigen recognizing;b. formation of endosome;c. ADC transferring from early endosome to lysosome;d&e. cytotoxic payload exerts antiproliferative function;f. cytotoxic effect lead to apoptosis.[1]
Introduction of Val-Cit-MMAE Linker
Because Phe-Arg dipeptide moiety is a general substrate of cathepsins B and L, this simple sequence was optimized to be Val-Cit which is specifically sensitive to cathepsin B. For screening a better cathepsin B substrate sequence, Citrulline was investigated as an isosteric and isoelectronic but less basic candidate of Arginine. In addition, Val-Cit showed better cleavage rate compared to other P1-Cit sequences among hydrophobic amino acids [3]. Concern that sterically demanding drugs such as DOX would be excluded from the active site of cathepsin B or would otherwise interfere with dipeptide binding led researchers to incorporate a self-immolative PABC spacer[4], which hydrolytically decomposes upon diacylation, spontaneously releasing the free drug.
MMAE(monomethyl auristatin E) is structurally related to dolastatin 10, a pentapeptide natural product that has been the subject of several human clinical trials for cancer therapy. Molecules in this family exert potent antitumor activities by inhibiting tubulin polymerization, and may also cause intratumoral vascular damage[5]. Among a diverse panel of 39 human tumor cell lines, none of the cell lines was resistant to AE, and the drug(average half-maximal inhibitory concentration(IC50) 3.2 ± 0.51 nM, 1 h exposure) was much more potent than other antimitosis agents such as vinblastine(average IC50 166 nM) and doxorubicin(average IC50 631 nM)[6]. To introduce AE onto antibody, the dimethylamino group was replaced by monomethylamino group which could be coupled to PABC spacer conveniently (Figure 2).
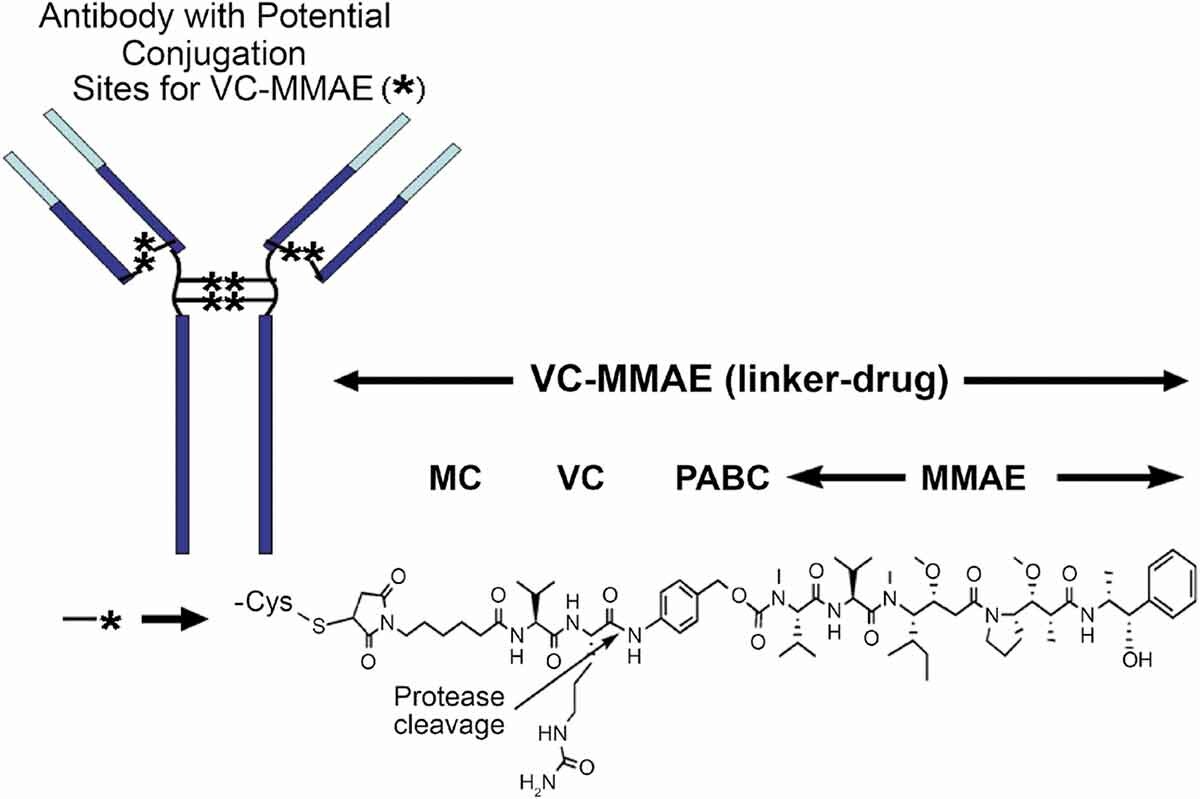
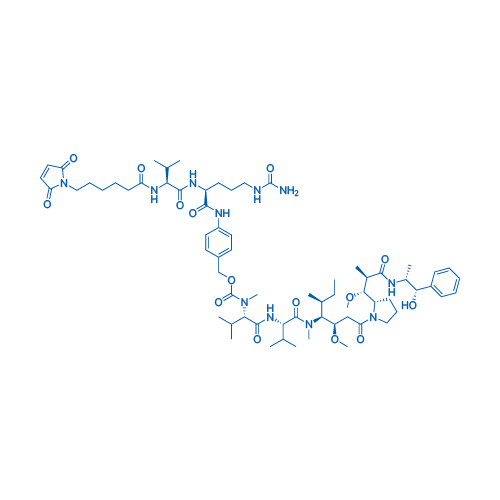
Figure 2: Chemical structure of a vc-MMAE ADC [7]
Clinical efficacy and application of vcMMAE
Genetech reported and compared the PK characteristics of the eight vc-MMAE ADCs using data from eight first-in-human (FIH) Phase 1 studies in cancer patients[7]. As a result, four ADC molecules(acMMAE) show that the exposure strongly correlate with efficacy(objective response rate, ORR)(Figure 3), which suggests their positive clinical efficacy.
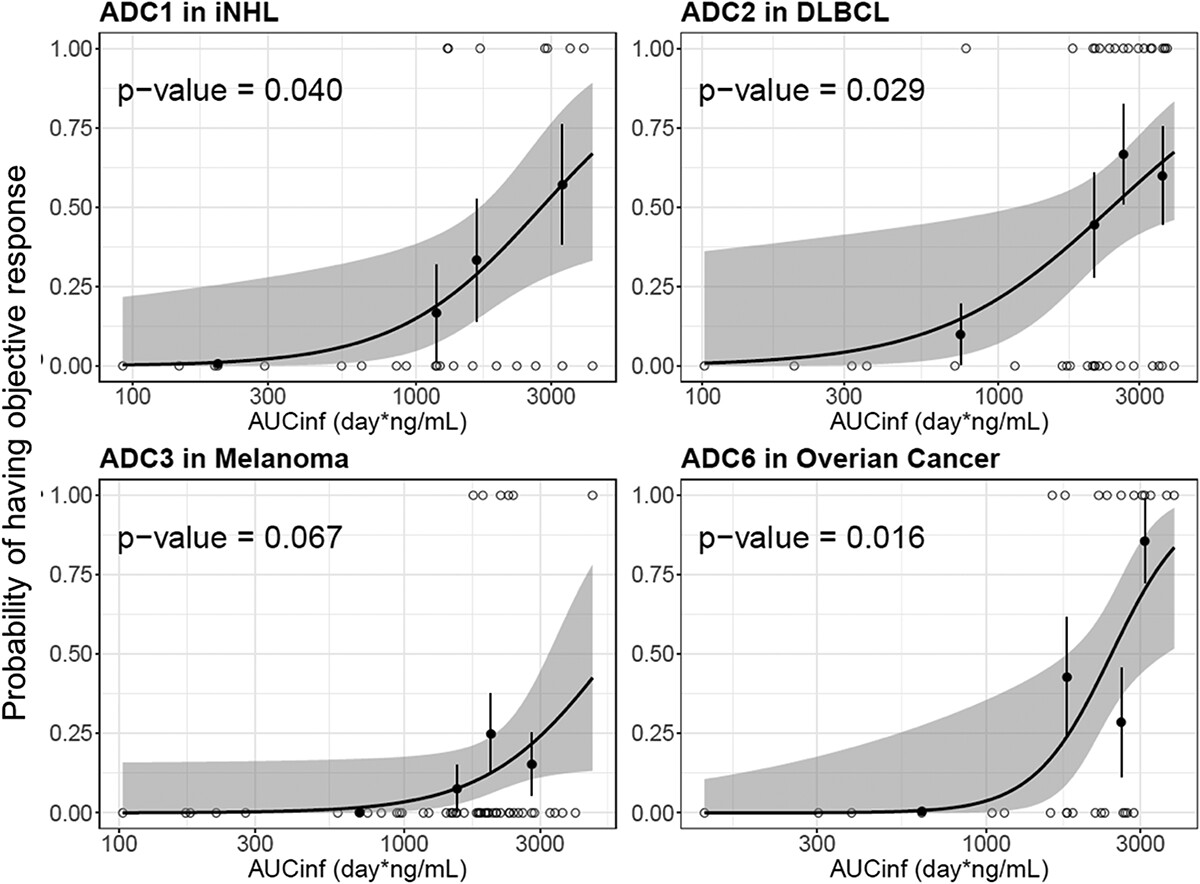
Figure 3: Logistic regression analysis of exposure-efficacy (ORR) response relationship by acMMAE AUCinf at Cycle 1.[7]
Recently, more and more ADCs which passed clinical test adopt vcMMAE as linker-payload combination, including Disitamab Vedotin of Remegen(indication: HER2+ gastric carcinoma/UC), Padcev of Seagen/ Astellas(indication: UUTUC), Tivdak of Seagen/Genmab(indication: cervical carcinoma), Adcetris of Seagen/Takeda(indication: HL/sALCL), Polivy of Roche(indication: DLBCL). In brief, vcMMAE was widely accepted in ADC design.
References
[1]Sievers, E. L.; Senter, P. D., Antibody-Drug Conjugates in Cancer Therapy. Annual Review of Medicine 2013, 64 (1), 15-29.
[2]Erickson, H. K.; Widdison, W. C.; Mayo, M. F.; Whiteman, K.; Audette, C.; Wilhelm, S. D.; Singh, R., Tumor Delivery and In Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody−Maytansinoid Conjugates. Bioconjugate Chemistry 2010, 21 (1), 84-92.
[3]Dubowchik, G. M.; Firestone, R. A.; Padilla, L.; Willner, D.; Hofstead, S. J.; Mosure, K.; Knipe, J. O.; Lasch, S. J.; Trail, P. A., Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific In Vitro Anticancer Activity. Bioconjugate Chemistry 2002, 13 (4), 855-869.
[4]Carl, P. L.; Chakravarty, P. K.; Katzenellenbogen, J. A., A novel connector linkage applicable in prodrug design. Journal of Medicinal Chemistry 1981, 24 (5), 479-480.
[5]Otani, M.; Natsume, T.; Watanabe, J.-i.; Kobayashi, M.; Murakoshi, M.; Mikami, T.; Nakayama, T., TZT-1027, an Antimicrotubule Agent, Attacks Tumor Vasculature and Induces Tumor Cell Death. Japanese Journal of Cancer Research 2000, 91 (8), 837-844.
[6]Doronina, S. O.; Toki, B. E.; Torgov, M. Y.; Mendelsohn, B. A.; Cerveny, C. G.; Chace, D. F.; DeBlanc, R. L.; Gearing, R. P.; Bovee, T. D.; Siegall, C. B.; Francisco, J. A.; Wahl, A. F.; Meyer, D. L.; Senter, P. D., Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nature Biotechnology 2003, 21 (7), 778-784.
[7]Li, C.; Zhang, C.; Li, Z.; Samineni, D.; Lu, D.; Wang, B.; Chen, S.-C.; Zhang, R.; Agarwal, P.; Fine, B. M.; Girish, S., Clinical pharmacology of vc-MMAE antibody–drug conjugates in cancer patients: learning from eight first-in-human Phase 1 studies. mAbs 2020, 12 (1), 1699768.